Contents
Base flipping
DNA-protein binding
The binding of DNA to proteins is important in many biological processes including translation, DNA replication, DNA repair. Some proteins bind to single-stranded DNA; others bind to double-stranded DNA. Different DNA-binding proteins target different parts of the DNA double-helix: the sugar-phosphate backbone is relatively exposed on the outside of the double helix, while the heterocyclic bases on the inside are hidden. When a protein needs to react with a single base within a DNA double helix, a mechanism must exist to make the base accessible to the enzyme: there must be a change in the local structure of the double helix around the target base. One can imagine several ways in which the double helix might become distorted to make the target base more accessible: bending or kinking, for example. DNA oligonucleotides are flexible molecules, capable of undergoing conformational changes. In some cases, the very nature of the double helix, with its canonical Watson-Crick base pairs, may be altered on binding to a protein; one remarkable example of this is base flipping.
Base flipping
Base flipping (or nucleotide flipping) was first observed in the interaction between the bacterial methyltransferase from Hha I and a 13-mer DNA duplex. In the X-ray crystal structure of the complex between enyzme and DNA one of the cytosine bases can be seen clearly to be rotated through 180° (from its Watson-Crick base-paired helical position) to a position on the outside of the double helix (Figure 1). In this position the cytosine base makes no interactions with the rest of the DNA duplex but is bound in the active site of the enzyme (the hydrogen-bonding and base-stacking interactions within the double helix are replaced by interactions with the protein).
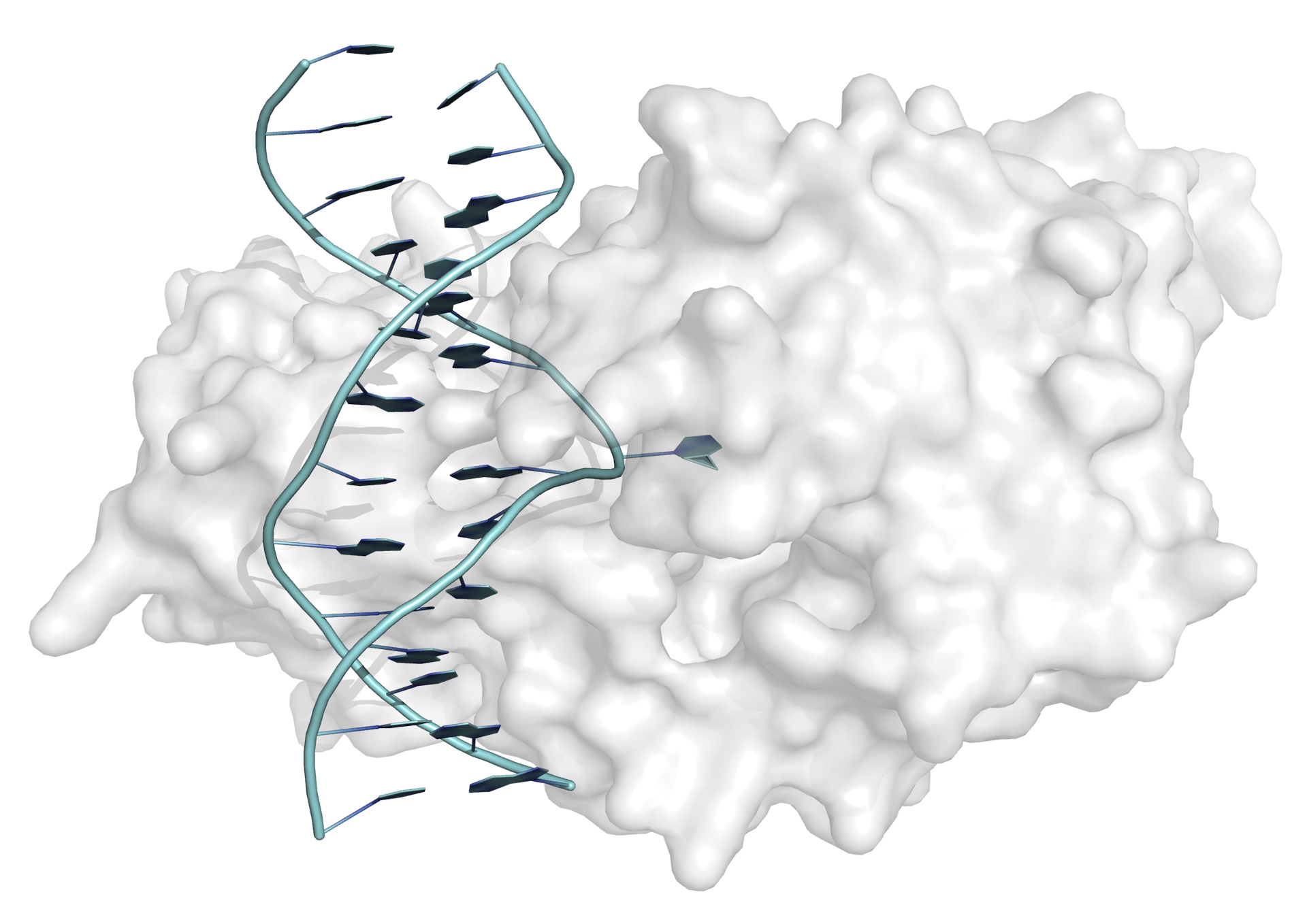
Once a complex is formed between enzyme and DNA, transfer of a methyl group can take place, to yield 5-methylcytosine. Following the methyl group transfer, the double helix is restored.
Since the discovery of base flipping in the complex with the methyltransferase from Hha I, base flipping has been observed in complexes with other enzymes that target a single DNA base, including other methyltranferases, glycosylases DNA repair enzymes and endonucleases, and base flipping may be important in other processes.
Kinetic and thermodynamic considerations
Thermodynamics
An equilibrium exists between the two (closed and opened) states of the DNA double helix (Figure 2). Under normal conditions the position of the equilbrium is heavily in favour of the closed state: for normal DNA, around 1 in 105 molecules of DNA are in the opened state at equilibrium.

The opened state can be stabilized by interaction with a host molecule (e.g. a DNA repair enzyme), which shifts the equilibrium in favour of the opened state. There is a large energetic cost in flipping one or two bases out of the double helix: hydrogen bonds between Watson-Crick base pairs are broken, base-stacking interactions are disrupted and torsional strain may be introduced in the backbone. Base flipping can only take place if there is an equivalent energetic gain when the DNA binds to the enzyme.
Kinetics
Base pair opening is rapid: the opening rate of a normal A·T base pair is approximately 1000 s−1, corresponding to a lifetime of only 0.1 μs for the opened state. If an enzyme is to react with the flipped state, it must do so before the DNA falls back to the closed state.
The role of enzymes in base flipping
The question arises whether enzymes play an active role in base flipping, or whether they simply bind to the base-flipped conformation that is present (in low concentrations) in solution. It is suggested that, because base-flipping always occurs in concert with some distotion (bending) of the DNA double helix (which assists rotation of the target base to its extrahelical position), base-flipping is an active process. Whether base-flipping is initiated by enzymes or by the presence of the flipped conformation in solution is unclear, and may vary for different classes of enzymes.
Damaged DNA, base flipping, and DNA repair
Not all DNA duplexes are equal: damaged DNA, containing mismatched base pairs, modified bases or lesions, is often less stable than analogous undamaged DNA (as indicated by a lower melting temperature, Tm). Damaged DNA has, therefore, a higher propensity to form an opened structure, with flipped-out base as the damaged site. DNA repair enzymes (such as DNA glycosylases) use this difference in stability of base pairs formed by damaged DNA as part of their strategy to recognise lesions.
Mechanism of base flipping in DNA glycosylases
Although base flipping was first observered in a methyltransferase, much of the recent work has been done on DNA glycosylases, for which base flipping has been studied in fine detail. DNA glycosylases repair damaged DNA as part of the base excision repair pathway, detecting lesions and excising them by breaking the N-glycosidic bond between sugar and base. Almost all DNA glycosylases employ base flipping to enable them to interact with and excise the target base.
Firstly, a nonspecific complex is formed between the DNA glycosylase enzyme and the double-stranded DNA, called the search complex (SC). Within the search complex, the association between enzyme and DNA is loose, and the enzyme is able to slide up and down the DNA chain. This sliding is interrupted every time the enzyme encounters a nucleotide, at which point a specific interrogation complex (IC) forms, in which the current base is flipped into an extrahelical position and bound in a secondary binding site of the enzyme (i.e. not the active site). This complex allows the enzyme to inspect the current base and determine whether it needs to be repaired. If a normal base is detected, the IC collapses to the SC; if a damaged base is detected, a third complex, the excision complex (EC) forms. In the excision complex, the flipped base is bound in the active site of the enzyme. The enzyme catalyzes the hydrolysis reaction, removing the damaged base from the DNA duplex. Once the reaction is complete, the search complex re-forms and the enzyme continues to the next base.
DNA glycosylases must be incredibly efficient to inspect all bases in the genome (3 × 109 base pairs) and detect and repair damaged bases (around 1 in 106 bases) before the genetic code is permanently altered (i.e. before the next cycle of DNA replication). Given that there are around 105 glycosylase molecules in the nucleus, each human glycosylase must sample around 70 000 base pairs of DNA in 12-24 hours (in practice, glycosylases can achieve this much faster).
The sliding mechanism, in which the enzyme and DNA do not diffuse away after each hydrolysis reaction but instead remain associated (in the search complex), allows the glycosylase to sample the flipped conformation that is present at low concentrations, and allows the enzyme to trap the flipped state during its short (∼0.1 μs) lifetime. Sliding also prevents undamaged DNA from acting as a competitive inhibitor of DNA repair enzymes, increasing the chances that all damaged sites are repaired.
Further reading
- Friedman and Stivers, Biochemistry 49 (24), 4957-4967, 2010.
- Stivers, Chem. Eur. J. 14, 786-793, 2008.