Contents
Contents
Nucleic acid-drug interactions
Introduction
Nucleic acids play an important role in cellular processes including cell division (DNA replication) and protein synthesis (transcription and translation). These processes occur both in healthy cells, and in cancer cells, in which case they are targets for anti-cancer drugs.
Small molecules may interact with or bind with DNA. Such compounds can be classified by their mechanism of action. The main classes of DNA binding molecules are:
- groove binders that sit in the minor groove;
- intercalators that sandwich between base pairs;
- alkylators that can chemically react with DNA, resulting in DNA alkylation; and
- DNA cleavage agents that have the ability to break DNA chains.
Each of these classes of molecules has a different structure and interacts with DNA in a different way.
Groove Binders
Minor groove binding molecules are usually constructed of a series of heterocyclic or aromatic hydrocarbon rings that possess rotational freedom. This allows the molecule to fit into the minor groove, with displacement of water.
Distamycin and netropsin
Distamycin and netropsin (Figure 1) are natural products possessing amido groups and, respectively, three and two N-methylpyrrole rings (distamycin can be denoted PyPyPy, and netropsin PyPy).

Distamycin and netropsin interact with AT-rich regions of DNA in the minor groove by forming hydrogen bonding and hydrophobic interactions (Figure 2). The terminal amidine group of the small molecule is basic, and serves to attract the drug molecule to the negatively charged DNA phosphodiester backbone. The 2-amino group of guanine prevents distamycin from binding to the minor groove of G·C base pairs by steric hindrance, thus conferring AT-selectivity on the drug molecule.
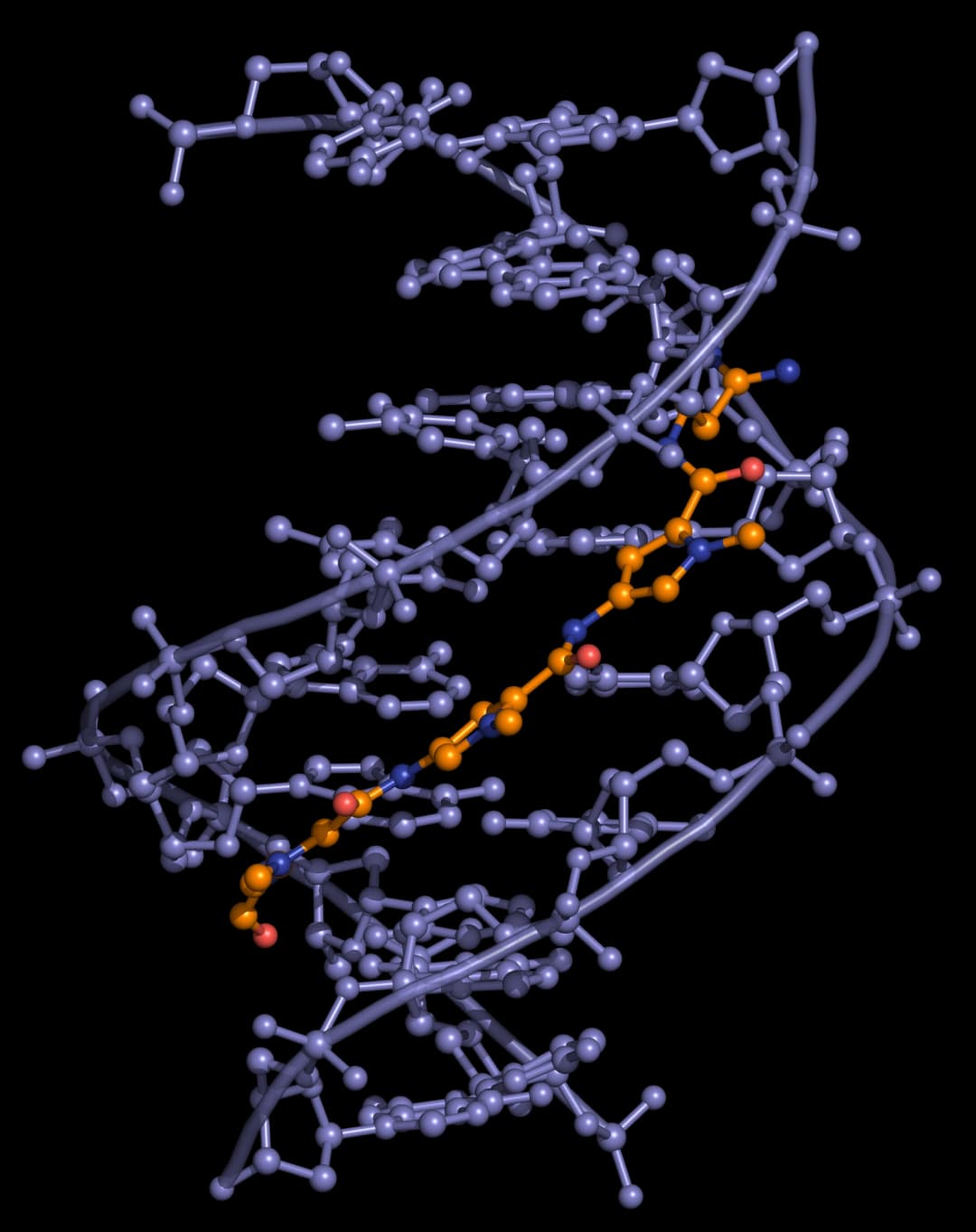
Lexitropsins
A series of dimers and trimers of distamycin and netropsin have been synthesized and studied in an attempt to increase the DNA binding region from 3 base pairs (monomeric drug) to 10 base pairs or more. These semi-synthetic compounds have been named Lexitropsins. As well as pyrrole rings (Py), some lexitropsins also incorporate imidazole (Im) rings. The ability to recognize specific DNA sequences of more than 10 base pairs would give rise to a powerful tool in molecular biology and antisense/antigene therapeutics (see Oligonucleotides as drugs), as this length of sequence starts to become meaningful in the context of the human genome.
Dervan polyamides
Unfortunately, simple oligomeric compounds like distamycin and netropsin do not have the ideal crescent shape to wrap around the minor groove of DNA, and they fail to recognize longer stretches of DNA. Dervan took this approach further in synthesizing a series of oligomeric "hairpin" polyamide molecules containing pyrrole and imidazole ring systems that are able to bind side-by-side in the minor groove of DNA with high affinity and in a sequence-specific manner. These molecules can be prepared by solid-phase methods.
Intercalators
Certain flat aromatic or heteroaromatic molecules can slide between the base pairs of DNA (intercalate) and stabilize the duplex without disrupting base pairing. Intercalation has the effect of lengthening the duplex by around 3 Å per bound drug molecule, causes unwinding of DNA, and prevents replication and transcription by interfering with the action of topoisomerases. The degree of unwinding depends on the structure of the intercalating molecule and the site of intercalation. The tight ternary complex formed between the intercalated drug, the DNA and the topoisomerase is lethal to proliferating cells, so intercalators are often more toxic to cancer cells than to normal cells.
Acridines

Acridines originated from the aniline dye industry and have been used as anti-malarial and antibacterial drugs. Amsacrine (Figure 3) is used in the treatment of leukemia and proflavine (Figure 3) was used in the Second World War to treat wounds. Proflavine contains amino groups that interact ionically with the negatively charged phosphates groups on DNA, whilst the aromatic ring system intercalates.
Polypeptides
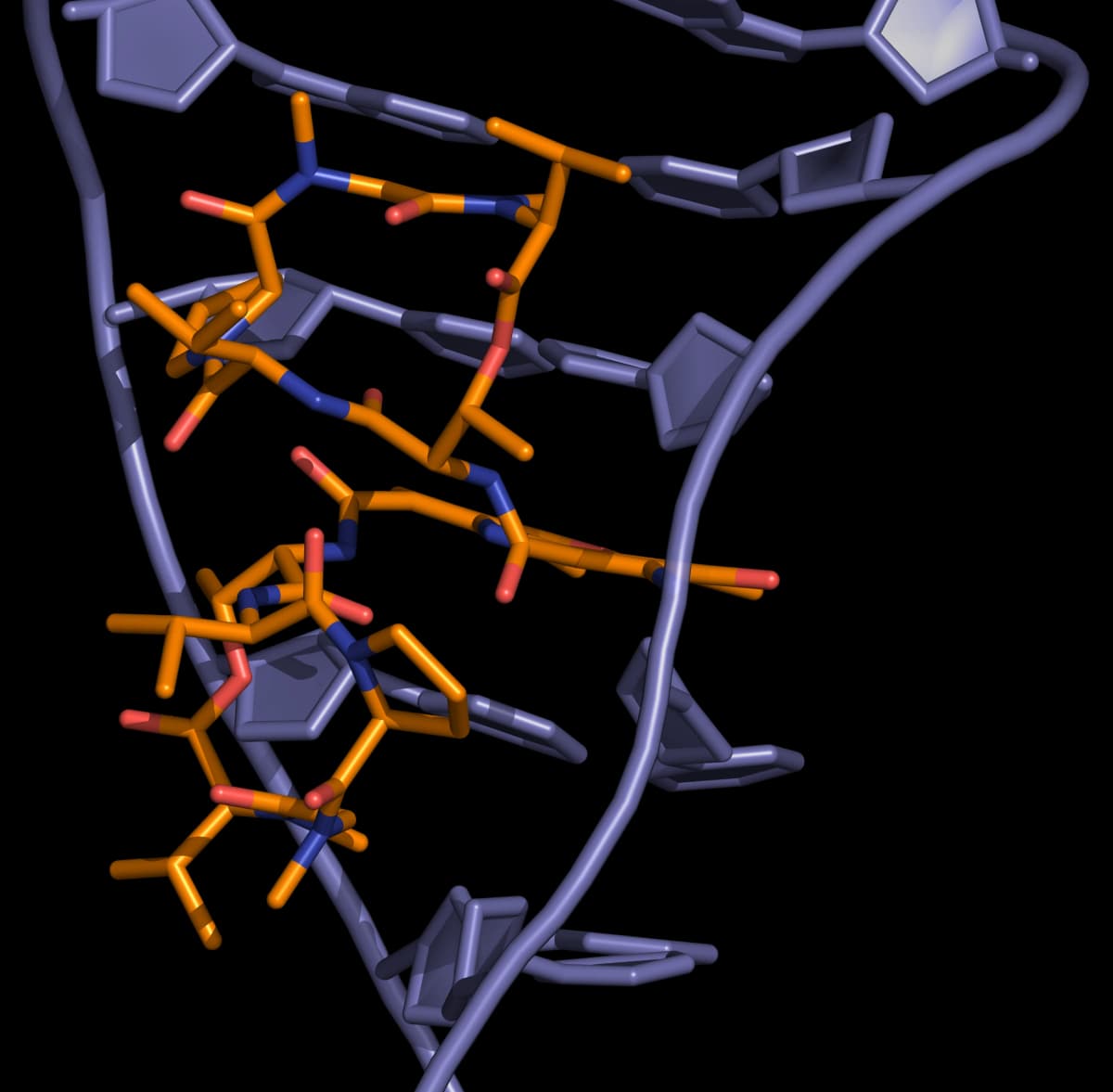
The Actinomycins (Figure 4) are polypeptide antibiotics isolated from Streptomyces strains.

Actinomycins inhibit both DNA synthesis and RNA synthesis by blocking chain elongation. They interact with G·C base pairs as they require the 2-amino group of guanine for binding. The phenoxazone ring slides into the double helix and intercalates, while the pentapeptide side chains interact with the DNA minor groove by forming hydrogen bonding and hydrophobic interactions. The result of these two mechanisms of interaction between small molecule and DNA (intercalation and minor-groove binding) is a very stable complex (Figure 5).
Anthracyclines
Doxorubicin (adriamycin) and daunorubicin (daunomycin), isolated from Streptomyces strains, are important examples of anthracycline antitumour antibiotics. Both possess an amino group on the sugar which, when protonated, forms an ionic interaction with the negatively charged DNA phosphate backbone. This bond helps to hold the molecule in place, allowing the planar aromatic ring system to slide into the double helix. Although doxorubicin and daunorubicin differ by only one hydroxyl group, they have different activities: daunorubicin is active only against leukaemias, but doxorubicin is active against leukaemias and a wide range of solid tumours.

Alkylators
Alkylators are strongly electrophilic compounds that react chemically with nucleophilic groups on DNA to form covalent bonds. The resulting DNA adducts are irreversible inhibitors of transcription and translation.
Nucleophilic substitution reactions at the DNA bases occur by both SN1 and SN2 mechanisms. The most reactive sites are those that are both nucleophilic and exposed in the grooves of the DNA duplex. The N(7) atom of guanine and the N(3) atom of adenine fulfil both of these criteria. Simple nucleophiles, for example ethyleneimines and methane sulfonates, tend to react via a SN2 mechanism, whereas the nitrogen mustards can form aziridinium ions that react via an SN1 mechanism.

There are several different classes of DNA alkylators, including nitrogen mustards, ethyleneimines, methanesulfonates, nitrosoureas, triazenes and cis platinum complexes.
Mustards
Sulfur mustard (Figure 8) is a highly toxic nerve gas that was used during the First World War. Although too toxic for use in cancer therapy, it led to the development of a series of compounds known as nitrogen mustards (Figure 8) that signalled the beginning of modern cancer chemotherapy. Mechlorethamine (Figure 8) was the first of these compounds to be used in the treatment of advanced Hodgkin's disease.

The mechanism of action of the mustards begins with the formation of an electrophilic aziridinium ion by displacement of chloride. The aziridinium ion is readily attacked by the nucleophilic DNA bases and the reaction sequence can occur a second time to cross-link two strands of DNA (Figure 9).

Mechlorethamine reacts principally with the N(7) atom of guanine, and quaternization of this nitrogen atom leads to scission of the N-glycosidic bond (Figure 10).

Mechlorethamine is a reactive molecule. It has a lifetime of only minutes in the body and is rapidly hydrolysed. Substitution of the methyl group with an electron-withdrawing aryl group reduces the nucleophilicity of nitrogen, slowing down the rate of aziridinium ion formation and therefore stabilizing the compound. However, such compounds are not sufficiently water soluble for intravenous administration. This problem has been solved by the use of carboxylate-containing aryl groups, resulting in the antitumour agent chlorambucil (Figure 11).

Ethyleneimines (aziridines)
Ethyleneimines are pre-formed aziridines and therefore constitute a natural extension of nitrogen mustards (Figure 12). To ensure antitumour activity, at least two ethyleneimine groups must be present in the molecule. To prevent protonation of the ethyleneimine, electron-withdrawing groups are attached (protonated ethyleneimines are too reactive). Lipophilic ethyleneimines are designed to enter the central nervous system.

Methanesulfonates
Methanesulfonates alkylate guanine at the N(7) position. Unlike nitrogen mustards, which tend to form inter-strand bridges, the methanesulfonates form intra-strand cross-links. An example is the bifunctional anti-cancer drug busulfan (Figure 13).

Platinum complexes
Cisplatin and derivatives
Platinum-based drugs are among the most widely used DNA-targeted agents used in cancer therapy. Cisplatin, and its derivatives carboplatin and oxaliplatin (Figure 14) represent a group of anti-cancer agents used in the intervenous treatment of testicular, melanoma, bladder, non-small-cell-lung cancers.

Cisplatin and its derivatives have substitution-active groups that, upon hydrolysis, establish strong covalent Pt(II)-N bonds with guanine and adenine bases, forming intra- or inter- strand crosslinks that trigger DNA structural changes, leading to cell death (Figure 15).

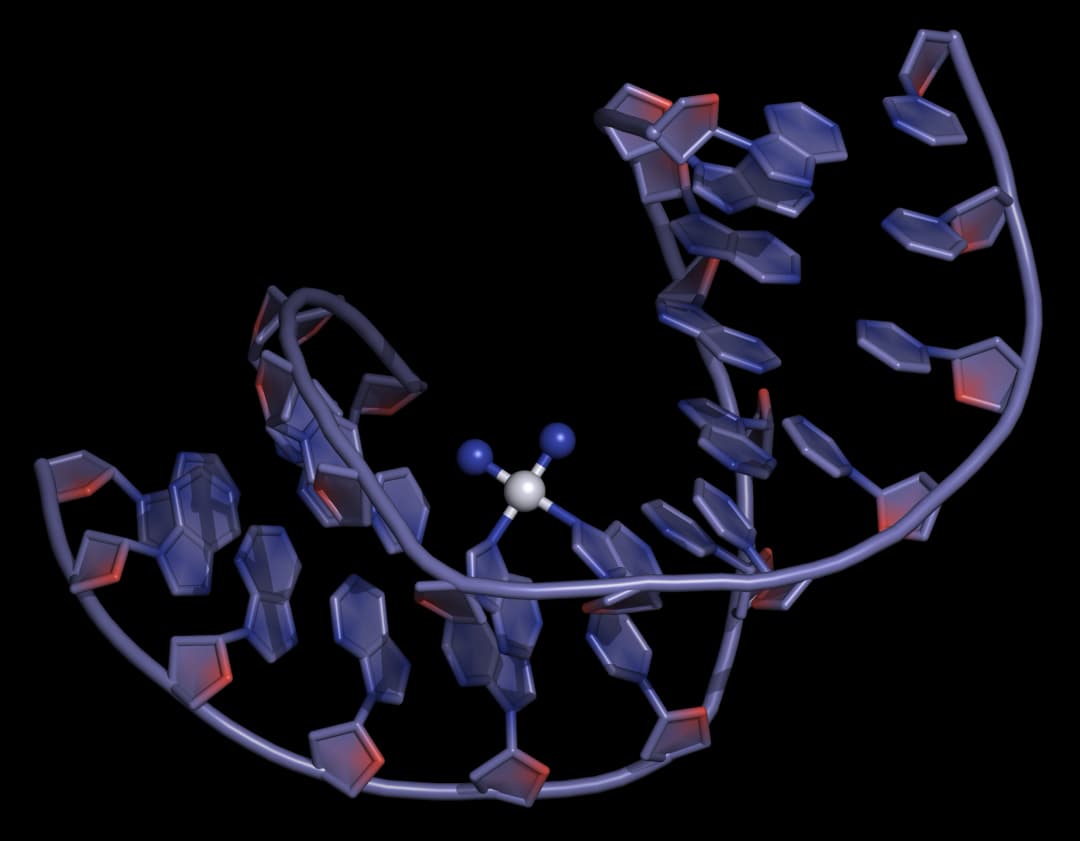
The trans isomer, trans-platin, is more susceptible to deactivation owing to its greater kinetic instability towards side reactions including hydrolysis, and lacks the correct symmetry for DNA cross-linking, and therefore has poor chemotherapeutic activity.
Despite the widespread clinical application of cisplatin-like drugs, resistance and dose-limiting toxicity issues have imposed limitations on therapeutic use, which has generated interest in the development of other, more targeted platinum drugs.
Polynuclear platinum complexes
One of a number of classes of platinum(II) compounds developed in response to the shortcomings of cisplatin and its derivatives, polynuclear platinum complexes have a distinct structure and exhibit a different mode of DNA binding. Polynuclear platinum complexes such as BBR 3464 (Figure 17), bind duplex DNA by forming long-range intra- and inter-strand crosslinks between purine bases up to 6 nucleotides apart; a mode of binding that is inaccessible to monomeric platinum complexes.

BBR 3464 made it to Phase II clinical trials for the treament of patients with various cancers, and work on it and related compounds is ongoing.
DNA cleavage agents
Bleomycin
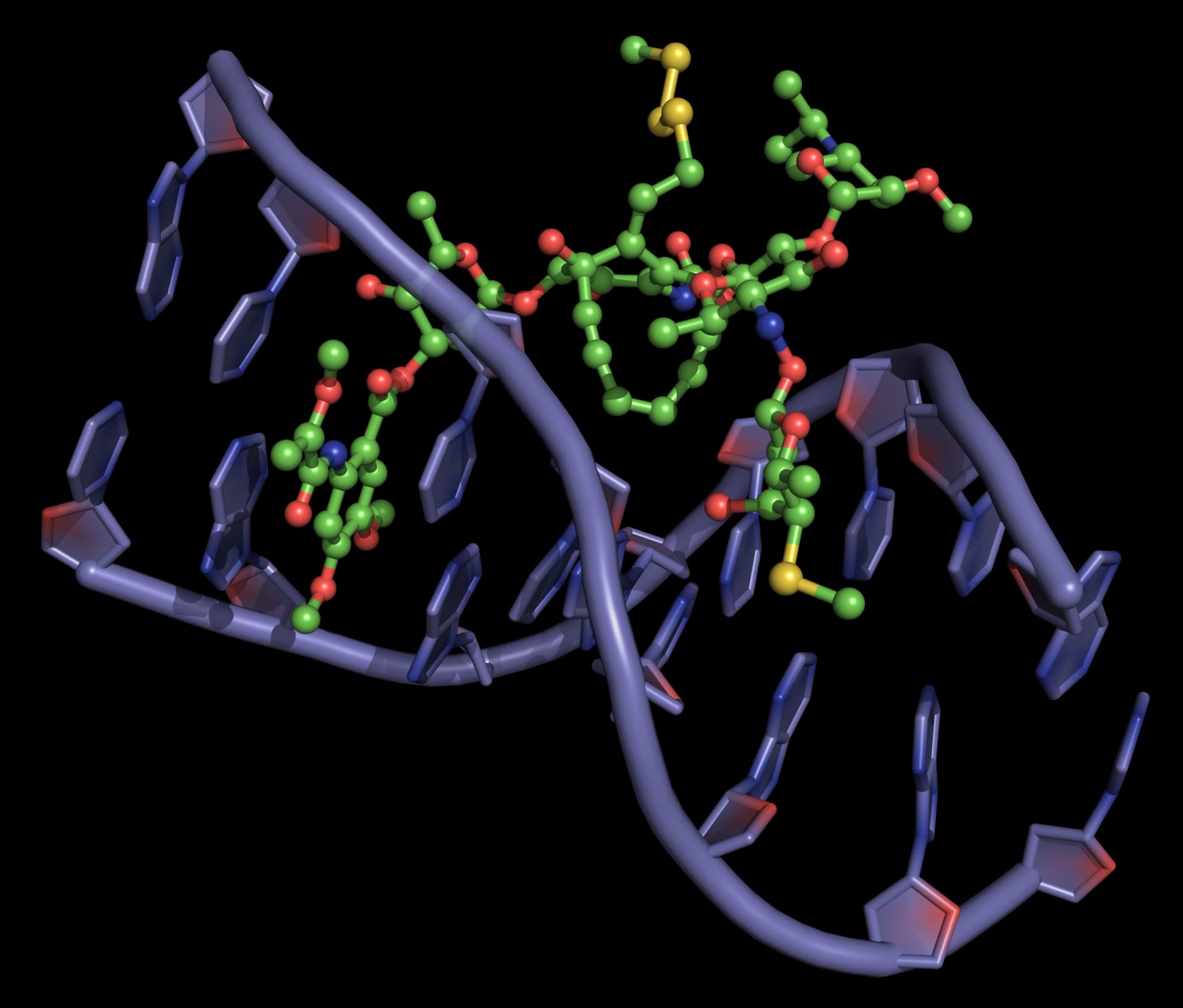
The classic DNA-cleaving anti-cancer antibiotic is bleomycin, a mixture of related antibiotics isolated from Streptomyces verticillus. Bleomycin is a glycopeptide antibiotic, the major component of which is bleomycin A2 (Figure 18).

There are three domains in bleomycin A2:
- Metal-binding domain: the pyrimidine, β-aminoalanine and β-hydroxyimidazole are involved in the formation of a stable complex with Fe(ii). Reaction with O2 gives a ternary complex believed to be responsible for the DNA cleavage activity.
- DNA-binding domain: the bithiazole moiety intercalates into the double helix and the attached side chain containing a sulfonium ion is attracted to the phosphodiester backbone.
- Carbohydrate domain: the gluose and carbamoylated mannose disaccharide are thought to be responsible for selected accumulation of bleomycin in some cancer cells. This domain does not appear to be involved directly in DNA cleavage.
Bleomycin binds tightly to guanine bases in DNA, particuarly in G-T and G-C-rich sequences. When the ternary complex of Fe(ii), bleomycin and oxygen attacks DNA, it abstracts hydrogen atoms. The resultant radicals react with oxygen to form peroxy species which then fall apart, resulting in chain cleavage.
Enediyne antitumour antibiotics
The enediyne antitumour antibiotics are anti-cancer compounds that cause the oxygen-dependent cleavage of the DNA phosphate backbone. They interact with the minor groove of DNA and then either a thiol or NADPH triggers a reaction that produces radicals. These radicals cleave the DNA chain. The major characteristic of these enediynes is the presence of a macrocyclic ring with at least one double bond and two triple bonds.
The two steps in antitumour activity are therefore
- activation of the enediyne
- action of the activated antitumour agent on DNA.
Neocarzinostatin
The first of the enediyne antitumour agents, neocarzinostatin (Figure 19), was isolated in 1965. Further enediynes, principally esperamicins and calichemicins, were not isolated until many years later.

Neocarzinostatin contains an active naphthoate ester which intercalates in DNA, positioning the diyne in the minor groove. Activation with a thiol followed, by a Bergman rearrangement, produces a diradical that is somewhat different from those produced by other enediynes (it is not a 1,4-dehydrobenzene diradical, but it is thought to behave similarly). The reactive diradical effects DNA strand scission by reaction with the C4'- and the C5'-atoms of the deoxyribose sugar, usually at deoxyadenosine and thymidine residues, consuming one equivalent of O2 per strand break (Figure 20). As neocarzinostatin is the oldest member of the enediyne class of antitumour agents, it is the most thoroughly studied.

Esperamicins and calichemicins
The esperamicins and calichemicins are similar to neocarzinostatin, having a bicyclo [7.3.1] ring and an allylic trisulfide attached to the bridgehead carbon, a 3-ene-1,5-diyne as part of the macrocycle and an α,β-unsaturated ketone in which the double bond is at the bridgehead of the bicyclic system (Figure 21).

The proposed mechanism of activation of the esperamicins and calichemicins begins with reduction of the trisulfide to a thiolate by NADPH or thiols in the cell. This results in an intramolecular Michael addition, which followed by a Bergman rearrangement gives the 1,4-dehydrobenzene diradical, the activated form of the esperamicin or calichemicin (Figure 22).

Enediynes interact with the DNA minor groove, so that the pro-radical centers of the enediyne moiety are positioned close to the proton abstraction sites (Figure 23).
Dynemicin A
Dynemicin A (Figure 24) combines structural features of both anthracyclines and enediynes. Dynemicin A binds to DNA by a combination of intercalation and minor groove binding. Dynemicin A can be activated by NADPH, thiols or light, and, once bound to DNA, can cause cleavage of one or both strands of the DNA helix.
